
Leveraging Ometa Flow for in vitro and in silico library generation
Leveraging Ometa Flow for in vitro and in silico library generation
Feature annotation is one of the major hurdles in analyzing and interpreting metabolomics data. A single untargeted metabolomics sample can produce tens of thousands of metabolic features, most of which do not match existing spectral libraries. While molecular networking can greatly expand the annotation of unknown molecules, increasing the number of compounds in your spectral libraries is one of the best ways to consistently increase the annotation rate of untargeted metabolomics data.
Traditionally, candidate compounds were synthesized individually before a reference spectra of a single pure compound could be collected and added to a spectral library. However, with millions of unknown compounds in public mass spectrometry data, compound synthesis remains a major bottleneck for improving annotation rates. The methods outlined here demonstrate how to rapidly expand spectral libraries both in vitro - and in silico - without the need to synthesize and purify single compounds. Step by step instructions on how to use Ometa Flow to create your own spectral libraries can be found in the accompanying technical documentation.
Creating spectral libraries in vitro using reverse metabolomics
A recent paper by Gentry et. al. outlines the process of creating spectral libraries for entire classes of novel compounds using reverse metabolomics.1 Reverse metabolomics takes the discovery process for novel molecules and turns it on its head: instead of measuring a novel compound in a biological sample and going through the difficult process of structure elucidation and synthesis, novel compounds are first chemically synthesized in vitro before they are identified in biological samples. In this case, the authors used combinatorial synthesis to generate many possible bile acid amide conjugations in a single synthesis reaction. Bile acids are a class of molecules with a handful of basic core structures conjugated to a variety of modifications.

Bile acid core structure and modifications
The core structures differ in their number of hydroxyl groups - for instance, a trihydroxylated bile acid will have a three -OH group attached to its core ring structure. The authors combined 8 bile acid core structures with 22 amino acids to create 176 possible bile amidates and then ran MS/MS on the entire reaction to create reference spectra for all synthesized molecules.
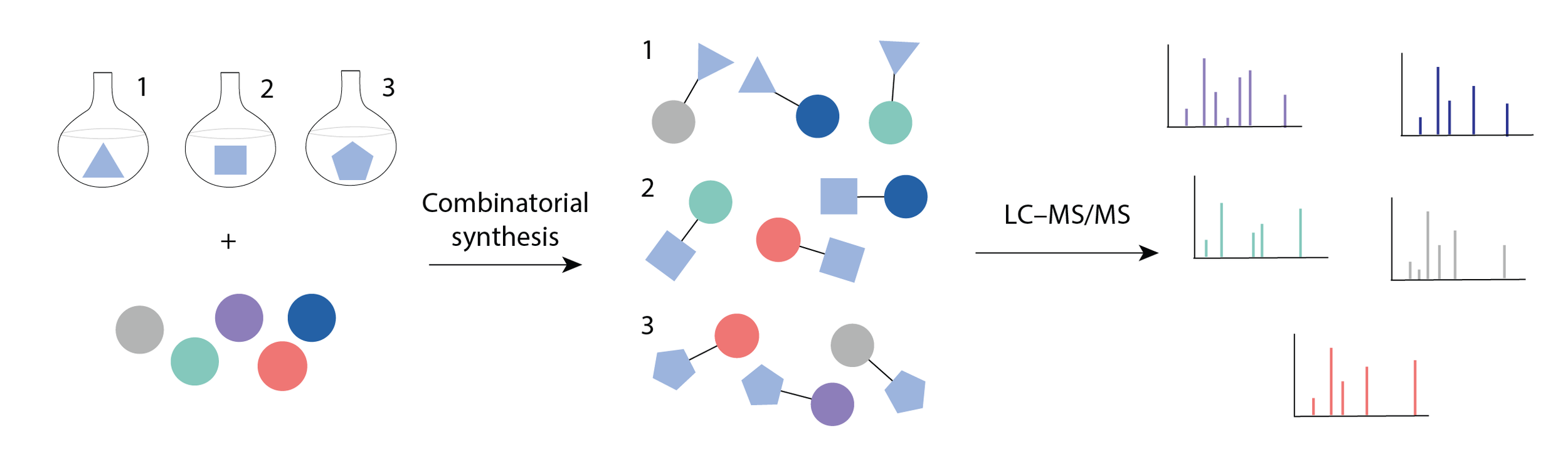
Combinatorial synthesis workflow. Figure adapted from Gentry et. al.
This strategy produced over 170 reference spectra of novel bile acid amide conjugations:

Bile acid-amide conjugation scheme. Figure adapted from Gentry et. al.
However, just because a chemical can be synthesized doesn’t mean it has biological relevance. To investigate whether these chemicals existed in vivo, Gentry et. al. employed the mass spectrometry search tool MASST to find their synthetic molecules in public data. MASST searched over 1.2 billion public spectra to find samples which contained the synthetic molecules. By pairing this information with metadata from ReDU, the authors were able to pinpoint species, organs, and even phenotypes associated with these novel molecules.
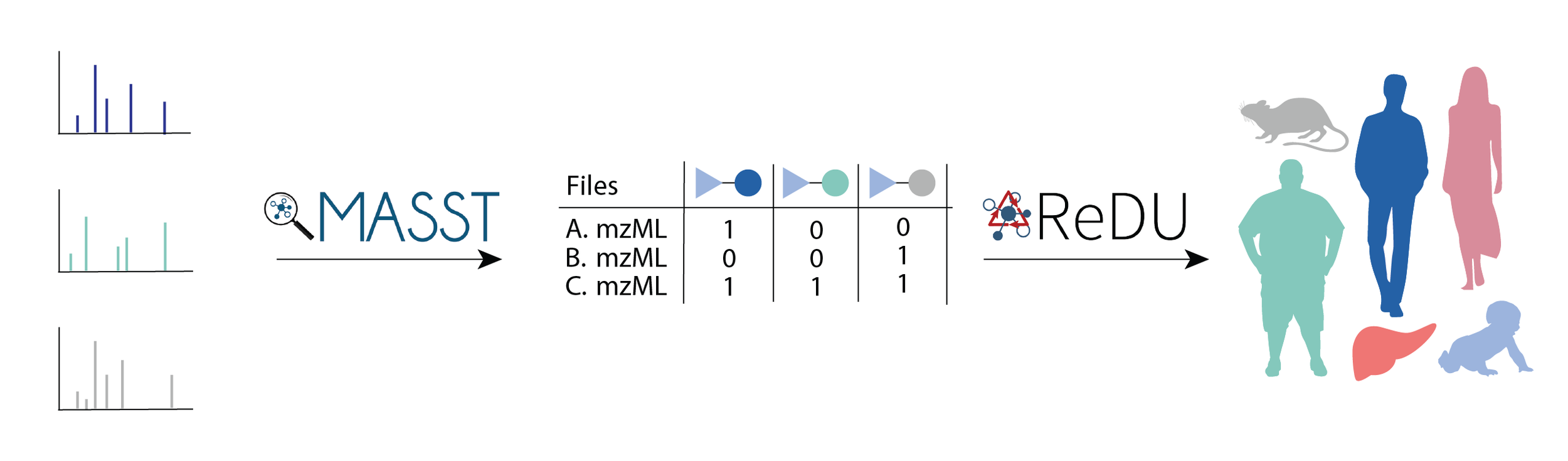
Figure adapted from Gentry et. al.
Creating spectral libraries in silico
Reverse metabolomics can rapidly expand spectral libraries, but it still relies on successful synthesis reactions to create reference spectra. This will limit the generation of spectral libraries to classes of compounds that are amenable to combinatorial synthesis.
In their recent paper, Mohanty et. al. used the mass query language MassQL to create in silico libraries for thousands of novel bile acids without a single synthesis reaction.2 Using spectra of known bile acids, the authors identified several diagnostic peaks for the bile acid core structure.

a. Representative spectrum of the trihydroxylated bile acid Glycocholic acid. Two peaks representing the bile acid core are highlighted in purple. b. MassQL query used to search for spectra containing those two representative peaks. An English translation of the query is also provided. Figure adapted from Mohanty et. al.
The authors then created MassQL queries to search for representative peaks corresponding to mono, di, tri, tetra, and pentahydroxylated bile acids. They used these queries to search the entire public MS/MS repository, identifying 594,365 spectra with those representative peaks. Using the known mass of the core structure, the authors were able to calculate the mass of each unique conjugation to the bile acid core (the delta mass). The figure below shows the number of delta masses corresponding to each bile acid core, and highlights the delta mass of 57.02, which was one of 15 delta masses that were found to be conjugated to all five bile acid cores.

Upset plot with number of modifications found on each bile acid core. Figure adapted from Mohanty et. al.
The reverse metabolomics technique described in the previous section increased the number of known bile acids from dozens to over 200. The MassQL search resulted in the identification of 5734 unique bile acid modifications in over 45,000 public samples. The authors were even able to pinpoint novel bile acids associated with specific species or health conditions. To confirm their presence in a variety of species, they synthesized and ran standards for a handful of these novel molecules.
Case study: annotation of novel bile acids in public data
To highlight how in-vitro and in-silico libraries can improve annotation of unknown molecules, the Ometa Labs team analyzed a publicly-available dataset containing fecal samples from volunteers with omnivore or vegan diets.3 The addition of the reverse metabolomics library more than doubled the number of bile acid annotations, while the in-silico library increased potential annotations over 10-fold.
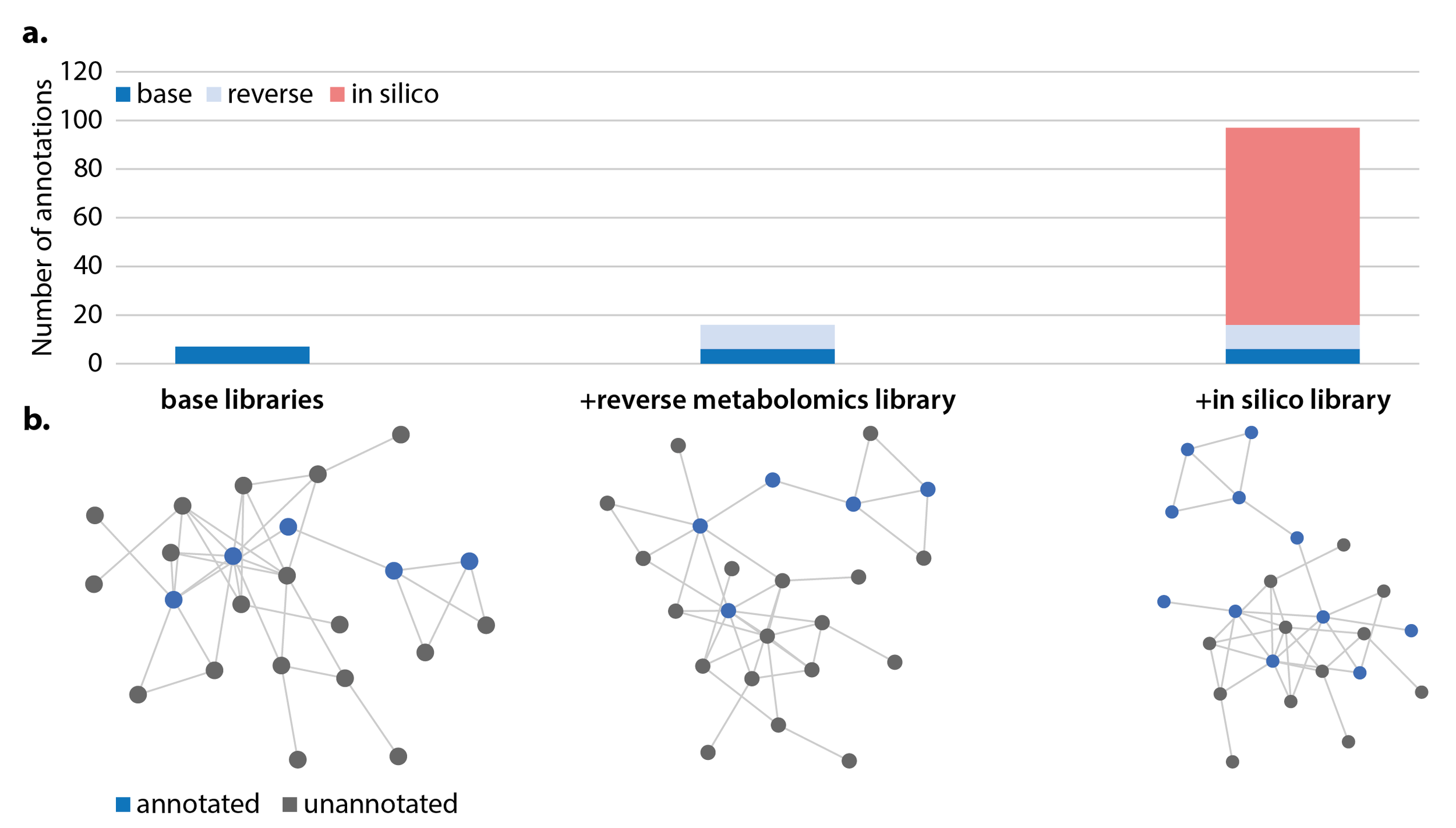
a. Number of bile acid annotations using base libraries, reverse metabolomics library, and in silico library. b. A representative subnetwork containing bile acids. Blue nodes represent a library match.
While in silico libraries can never provide the same level of confidence as a match to a synthesized reference standard, they can prove an invaluable starting point for further discovery. Both the reverse metabolomics and in silico bile acid libraries described above are now available to all Ometa Flow users. Step-by-step instructions on creating these libraries for yourself with Ometa workflows can be found in the accompanying technical documentation.
References
- Gentry, E. C. et al. Reverse metabolomics for the discovery of chemical structures from humans. Nature 1–8 (2023) doi:10.1038/s41586-023-06906-8.
- Mohanty, I. et al. The underappreciated diversity of bile acid modifications. Cell 0, (2024).
- MSV000086989, accessed through MassIVE.